
 Fact-checked by
Fact-checked by 
ALS stages and progression timeline
Amyotrophic lateral sclerosis (ALS) is a neurodegenerative disorder marked by progressively worsening stages of progression. People with ALS experience muscle weakness and neurological dysfunction, but initial symptoms are typically mild and affect only one part of the body. As the disease becomes more and more severe, these issues gradually spread to the entire body, affecting a range of bodily functions.
There can be a lot of variation in the order in which ALS symptoms appear and how quickly the disease progresses. However, as ALS advances and makes it more difficult to perform daily activities, patients go through several stages of progression.
How many stages of ALS are there?
There are several formal scales designed to determine ALS staging. The two most commonly used are the King’s College staging system, which assesses the spread of the disease throughout the body, and the Milano-Torino (MiToS) system, which measures how symptoms impact a patient’s ability to perform certain functions.
Yet, according to the Muscular Dystrophy Association, the disease can be more generally divided into four stages, including:
- early stage
- middle stage
- late stage
- end stage.
No available treatment options can prevent patients from progressing through the multiple stages of ALS. The timing of each stage may vary substantially from patient to patient, and treatment may extend some stages by a few months, but the vast majority of individuals with ALS will go through these stages over a period of about two to five years.
Early-stage ALS
In the earliest stage, the symptoms of ALS are usually quite mild and affect one region of the body. In about two-thirds of patients, the disease initially starts in the limbs, usually affecting muscles in the hands, feet, calves, and forearms. This form of the disease, known as limb-onset ALS, commonly manifests with problems such as:
- trouble with fine motor tasks like writing or buttoning clothes
- difficulty gripping objects
- balance issues
- problems with walking such as frequent tripping.
The other third of patients have bulbar-onset ALS, and their first symptoms result from weakness in the bulbar muscles — that is, the muscles around the mouth and throat. This can lead to:
- problems with articulation and slurring speech
- an unusually hoarse or quiet voice
- difficulty swallowing or chewing.
Other ALS early signs may include:
- muscle stiffness, twitching, and cramping
- unusual fatigue
- loss of muscle bulk (atrophy).
The majority of patients retain a fair amount of functionality and independence during the early stage of ALS, which typically lasts about a year. But because it often takes many months for patients to get a formal ALS diagnosis, many have already progressed past the early stage by the time the disease is confirmed.
For patients who are still in the early stages of ALS when they are diagnosed, medical care mainly focuses on establishing a treatment plan, including making decisions on medications and supportive therapies, and planning ahead to make accommodations and preparations for later disease stages.
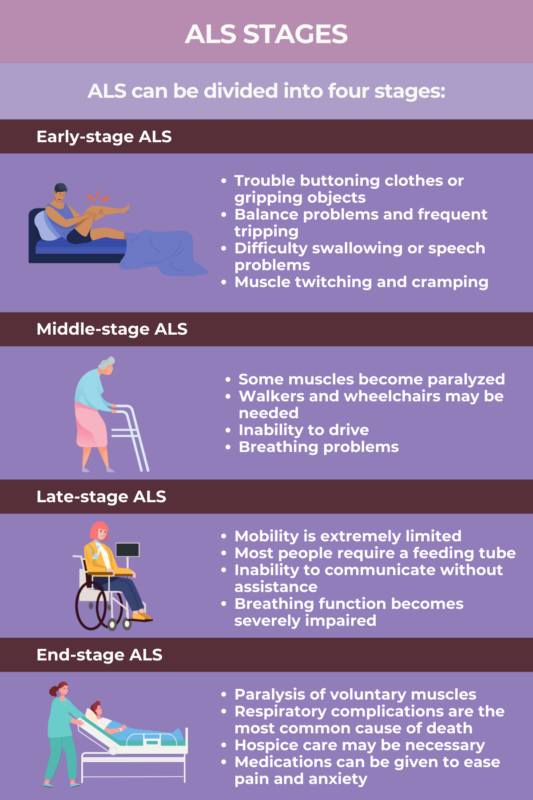
Middle-stage ALS
As ALS progresses into the middle stage, muscle weakness becomes more severe and spreads, affecting more regions of the body. In this stage of disease:
- Some muscles become completely paralyzed and others are weakened, though some muscles may remain unaffected.
- Cramping and twitching may occur, and the loss of muscle mass (atrophy) intensifies.
- Muscles that are no longer moving may become shortened and hardened, known as contractures, which may cause pain and rigidity around joints.
- Weakness in the mouth and throat can cause difficulty speaking, as well as problems eating and managing saliva, which can increase the risk of choking.
- Weakness of muscles in the chest may cause difficulty breathing, particularly when lying down.
- Some patients experience pseudobulbar affect, which is characterized by bouts of uncontrolled laughing or crying that are not related to a person’s true emotions.
Patients in the middle stage of ALS usually retain the ability to move some parts of their body under their own power, but ALS-related changes will begin to hamper mobility in this stage. That means that most patients will require some assistance with day-to-day activities, with that need increasing over time. Falls may become more common, and patients are typically unable to stand unassisted after a fall.
In this stage, patients usually are unable to drive, and they generally will rely on aids like walkers or wheelchairs to help them get around. Medical interventions like feeding tubes and respirators are often needed to help patients get enough nutrition and maintain breathing function.
The middle stage of ALS may last anywhere from a few months to more than a year.
Late-stage ALS
By the late stages of ALS, most muscles involved in voluntary movement become paralyzed. Patients generally have very limited or no ability to move under their own power, and will require assistance with most day-to-day activities. At this stage:
- Eating and drinking by mouth may not be possible; patients often get their nutrients via feeding tube.
- Speech may be difficult or impossible, and patients may need to rely on alternative methods for communication.
- The muscles of the chest are severely weakened, causing difficulty breathing and increasing the risk of pneumonia.
- Low oxygen levels due to poor breathing can contribute to symptoms like fatigue, fuzzy thinking, and headaches.
Most individuals in late-state ALS will require ventilatory support when breathing function declines significantly.
Exact timelines vary, but most people with ALS will be in the late stage within two to three years after they initially start developing symptoms.
End-stage ALS
ALS is a fatal disease, and most people with the disorder will die within about two to five years after symptom onset. The most common cause of death in ALS is respiratory failure, when breathing ability is compromised to the point that the body cannot get enough oxygen to stay alive.
Less common causes of death in ALS may include:
- malnutrition as a result of swallowing issues
- a blockage in the lung’s blood vessels, known as pulmonary embolism
- problems with heart rhythms, known as cardiac arrhythmias
- pneumonia due to food or liquid getting into the lungs.
In the final stages of ALS, medical care focuses mainly on maximizing the patient’s comfort. Medications may be given to help relieve pain and ease anxiety.
Hospice care — a form of end-of-life care that aims to help meet the physical, emotional, and spiritual needs of the person with ALS and any family members and close friends — may be given at home or in a specialized care facility. Patients typically will not require hospital or emergency care.
Patients and families should plan ahead for the end stage of ALS to ensure their end-of-life care is consistent with their wishes. It is recommended that hospice is contacted early to understand what services are available at home or in a facility, and determine what will best suit the patient and family members when that stage is reached. Available services and care options may depend on the patient’s geographical location.
ALS progression timeline
There can be substantial variation from person to person in how quickly ALS progresses. While the average survival time among people with ALS is up to five years from the onset of the disease, about 1 in 5 patients will live at least five years, and 1 in 20 will survive two decades or longer.
While there is no cure for ALS, and no therapies can reverse symptoms that have already accrued, several treatments can help to slow the decline in function over time for people living with ALS. Some also can extend survival by a few months.
This means that ALS stages will have variable duration among patients.
While it’s difficult to determine how long patients will remain in each of the four general stages of ALS disease progression, studies examining more formal disease stages can provide a general idea.
One of the formal scales more commonly used in studies to document ALS progression is the King’s College staging system. This system assesses whether patients have substantial symptoms affecting four parts of the body: the upper limbs, the lower limbs, the bulbar muscles around the face, and the diaphragm. The diaphragm is a large muscle in the chest needed for breathing.
The system is staged from 1 to 4 based on how many of these regions have symptoms, with a fifth stage representing death.
- Stage 1, representing the early stage of disease, typically lasts from about nine months up to 1.5 years. In general, patients with limb-onset disease and those who receive ALS treatment early in the disease course will stay longer in this stage.
- Stages 2 and 3, which can be generally thought of as the middle stage of ALS, together last anywhere from about nine months to just longer a year for most patients.
- Stage 4, typically corresponding to the late stage, usually lasts about four to seven months for most individuals, though it may be longer depending on what kinds of life-supporting treatments are given.
- The time to stage 5, which can be seen as the duration of the end stage, is usually about three months for most patients.
The MiToS system is another commonly used formal scale that assesses whether patients have impairment in four main areas of functionality: movement, swallowing, communication, and breathing. It stages patients on a scale of 1 through 4 based on how many of these areas are impaired.
A stage 0, indicating no impairment, is included in the MiToS system, as is a fifth stage representing death from ALS.
As the King’s College and MiToS systems measure different but complementary information, they are often used in combination to help track patients’ progression.
ALS News Today is strictly a news and information website about the disease. It does not provide medical advice, diagnosis, or treatment. This content is not intended to be a substitute for professional medical advice, diagnosis, or treatment. Always seek the advice of your physician or other qualified health provider with any questions you may have regarding a medical condition. Never disregard professional medical advice or delay in seeking it because of something you have read on this website.
FAQs about ALS stages
Category:
Stages
Amyotrophic lateral sclerosis (ALS), also called Lou Gehrig’s disease, can be broadly divided into four stages: an early stage in which symptoms first appear, a middle stage where symptoms worsen and mobility becomes substantially impaired, a late stage characterized by near-total paralysis, and an end stage that leads to death.
Category:
Stages
The speed at which amyotrophic lateral sclerosis (ALS) progresses can vary a lot from person to person, but typically the disease progresses quite rapidly. Most patients reach the late stage of ALS within two to three years after symptom onset, and the average survival time is about two to five years from onset.
Category:
Stages
Although there is no cure for amyotrophic lateral sclerosis (ALS), there are several approved therapies that can slow disease progression, helping patients to maintain functionality for longer and prolonging survival. Patients and their healthcare team should establish a treatment plan to slow progression and help with symptoms early in the disease course.
Category:
Stages
The earliest signs of amyotrophic lateral sclerosis (ALS) vary from patient to patient, and depend on the region of the body that is first affected. For the vast majority of patients, in which limbs are first affected, early symptoms include difficulty grabbing objects or writing, and trouble with walking and balance. When the first muscles affected are located around the mouth and throat, initial symptoms include problems speaking or swallowing.
Category:
Stages
The final stages of amyotrophic lateral sclerosis (ALS), in which patients are fully paralyzed, typically last just a few months. However, the disease course will largely depend on the exact type of ALS and disease-associated mutations a person has, as well as on the kind of medical treatments and supportive therapies that are given.
Related Articles
-
-

-
 Discussion
Discussion
-

-

-
