
FDA advisory committee votes against NurOwn approval for ALS
17 to 1 vote says data do not support treatment's effectiveness
Written by |

In a nearly unanimous vote, an advisory committee to the U.S. Food and Drug Administration (FDA) said there’s not enough evidence to support the efficacy of the experimental stem cell therapy NurOwn as a treatment for amyotrophic lateral sclerosis (ALS).
The committee voted on a single question in its meeting: “Do the data presented demonstrate substantial evidence of effectiveness for treatment of mild to moderate ALS?”
In all, 17 of the 19 committee members voted no. There was one yes vote and one abstention.
An official decision from the FDA on NurOwn is set to be handed down in December. The regulatory agency is not obligated to abide by the committee’s vote, but it usually does.
“The discussion in today’s advisory committee meeting, and the heartrending testimony of those living with ALS and their loved ones, underscores not only the need for regulatory flexibility but also for continuing research in the field,” Chaim Lebovits, president and CEO of BrainStorm Cell Therapeutics, NurOwn’s developer, said in a company press release, adding, “The people of BrainStorm will do everything in our power to fulfill the obligation we deeply feel we owe to the ALS community, and in the coming weeks we will explore all options available to us.”

FDA found no evidence of so-called floor effect in trial
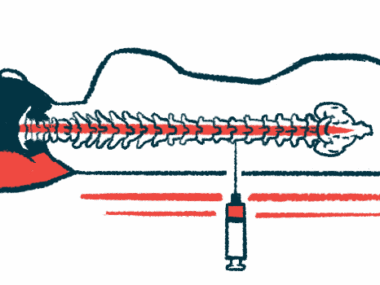
NurOwn is a cell-based therapy that involves harvesting stem cells from a patient’s bone marrow, engineering them in a laboratory so they secrete molecules that promote nerve health, and then infusing the cells back into the patient’s spine.
Following a decade of testing, BrainStorm submitted an application to the FDA last year seeking approval of NurOwn in the U.S. The agency initially refused to review the application, saying there was not enough evidence to support an approval.
However, but BrainStorm forced a new review and an advisory committee meeting using the FDA’s “file over protest” procedure.
BrainStorm’s application includes data from several clinical trials, including a large Phase 3 study (NCT03280056) that tested NurOwn against a placebo in 189 people with rapidly-progressing ALS.
The study aimed to show that NurOwn could slow the progression of the disease, as measured by the ALS Functional Rating Scale-Revised (ALSFRS-R), a standardized measure that assesses a person’s ability to perform activities of daily living. But final results showed no difference in progression between patients given NurOwn or a placebo.
The company argued that this might be due to a so-called floor effect — where some patients had ALSFRS-R scores so low that they couldn’t meaningfully get lower.
In post hoc analyses done once the study was over, the company analyzed only subsets of patients expected not to have a floor effect. Those results generally suggested slower disease progression with NurOwn compared with a placebo.
In briefing documents put out before the committee meeting, the FDA said it did not find evidence of a floor effect in the patients that BrainStorm had flagged. The agency also cautioned that post hoc analyses are heavily prone to false-positive results, and, overall, reiterated its stance that there are not enough data to support an approval of NurOwn.
The committee’s vote was a sad outcome for the ALS community, who have too few options to help manage this merciless and deadly disease.
The advisory committee has now strongly voted that the available data are not enough to demonstrate that NurOwn is effective in ALS — a decision that further supports a potential rejection by the FDA.
“The committee’s vote was a sad outcome for the ALS community, who have too few options to help manage this merciless and deadly disease,” said Stacy Lindborg, PhD, co-CEO of BrainStorm.
“We firmly believe that the totality of data presented for NurOwn today provide a compelling case for approval, with clinical evidence in those with less advanced disease supported by strong and consistent biomarker data that are predictive of clinical response,” Lindborg said.
Lindborg suggested that too little time was given during the meeting for BrainStorm to answer all of the committee’s questions.
“We truly did our best to make the NurOwn data clear to the FDA advisory committee. Unfortunately, had more time and opportunity been allowed, many remaining questions posed by advisory committee members could have been sufficiently addressed,” Lindborg said.


Leave a comment
Fill in the required fields to post. Your email address will not be published.